| Laboratory of Computational Proteomics Research: Proteomics |
|
Identification of proteins and characterization of their post-translational modifications.
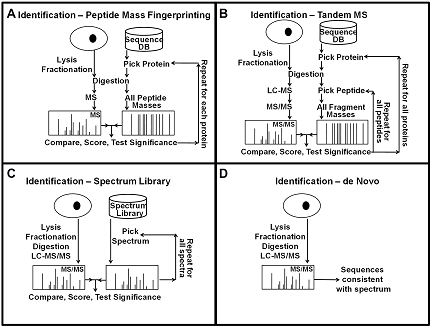
Mass spectrometry-based protein identification has become an invaluable tool for elucidating protein function, and several methods have been developed for protein identification, including sequence collection searching with masses of peptides or their fragments, spectral library searching, and de novo sequencing (Fig. 1). The first step in protein identification is to find peaks in the mass spectra that correspond to peptides and their fragments. It is important to find all the relevant peaks and at the same time minimizing the number of background peaks. This can be achieved by scanning the spectra for peaks of the expected width and selecting peaks above a signal to noise threshold, and then picking the monoisotopic peak for each isotope cluster. After picking the peaks, spectra with low information content that could not produce any meaningful results can be removed to increase the speed of subsequent analysis. In all mass spectrometry-based identification methods, a score is calculated to quantify the match between the observed mass spectrum and the collection of possible sequences. These scores are highly dependent on the details of the algorithm used, and they are not always easy to interpret because the interpretation of the score depends on properties of the data and the search results. Therefore, it is desirable to convert the score to a measure that is easy to interpret, such as the probability that the result is random and false. For this conversion, the distribution of random and false scores is needed. Estimates of this distribution can be generated using either simulations, collecting statistics during the search, or direct calculations.
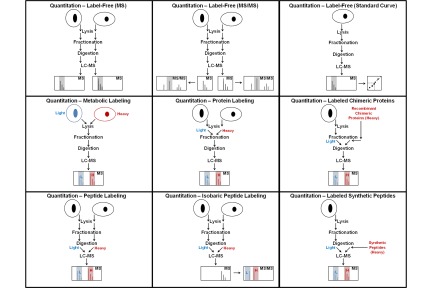
References Quantitation of proteins and peptides Mass spectrometry (MS)-based quantitative proteomics has been applied to solve a wide variety of biological problems, and several MS-based workflows have been developed for protein and peptide quantitation (Fig. 1). In mass spectrometric quantitation methods it is usually assumed that the measured signal has a linear dependence on the amount of material in the sample for the entire range of amounts being studied. A prerequisite for accurate quantitation is that unwanted experimental variations in sample extraction, preparation, and analysis be minimized, and it is therefore critical that each step in the workflow is optimized for reproducibility.
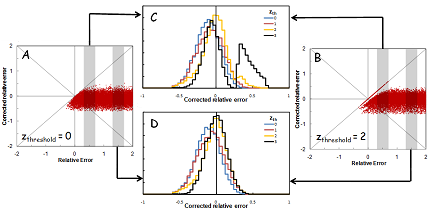
When quantitation of proteins in complex samples is based on the intensity of peptide precursor and fragment ions, interference can distort the measurements. It is important to detect and correct for these interferences. We used computer simulations as a tool to investigate the feasibility of correction for interference in MRM analyses. In our simulations, it was assumed that the expected relative intensity of the transitions for a peptide is known. Hypothetical interference was added to one or more transitions, and random noise was added to all transitions. The distribution of the noise was obtained from repeated measurements. Interference was detected by measuring the deviation of the intensity ratios of transitions from the expected ratios, and detecting outliers. The transitions with interference were removed and the peptide quantity was calculated using only the transitions without interference.
References
|